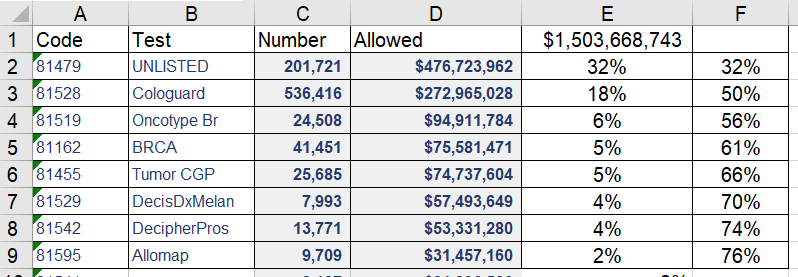
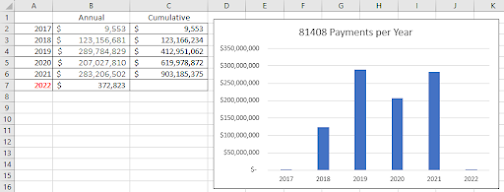
On October 18, 2023, Dr Scott Gottlieb, prior head of the FDA, provided a keynote address for the 13th Annual "
Value Based Cancer Care" conference in New York City.
In the first half hour, Dr. Gottlieb highlighted his upcoming book on the history and future of cell therapies. Next, an interview by John O'Brien of the National Pharmaceutical Council explored the challenges of reimbursement innovation, regulatory decision-making, and regulation of AI applications. These topics all emphasized the need for FDA adaptability in an evolving healthcare landscape.
####
I asked Chat GPT3.5 to write up an essay based my detailed conference notes today; I've edited it.
####
The 13th annual Value-Based Cancer Care conference kicked off with a powerful keynote address by Dr. Scott Gottlieb, the former Commissioner of the FDA and a healthcare policy expert. The hour included an interview by John O'Brien (National Pharmaceutical Council).
Dr. Gottlieb gave us a glimpse into his upcoming book, on the history and promise of CAR-T cell therapies. He highlighted situations where the FDA provided "regulatory tailwinds" that encouraged the development of cell therapies.
The journey of cell therapy (CAR-T therapy) in oncology has been marked by decades of incremental progress punctuated by significant breakthroughs. There was constant interaction between our growing knowledge of the cell biology of cancer, and the science and technologies of emerging cell therapies. Early progress was related to innovations in bone marrow transplantation, and the role of emerging role of T cells.
Dr. Gottlieb touched upon the transformative concept of engineering T cells to combat tumors, moving from passive harvesting and infusion to the idea of creating anti-tumor T cells. This led to the development of chimeric antigen receptor T cell (CAR-T) therapies.
Gottleib also highlighted the pivotal role of Dr. Carl June at the University of Pennsylvania Dr. June's work in stimulating and replicating T cells outside the body was a key part of bringing CAR-T therapies to healthcare, and the Penn group was equally interested in innovations to industrialize and scale the procedure. Gottlieb has never forgotten a meeting at Penn where June argued convincingly that the FDA (at the time) was hindering innovation and needed to be more adaptive.
One of the key takeaways from Dr. Gottlieb's talk was the importance of patience and long-term investment in medical research. He emphasized the critical role played specifically by physician-scientists who are committed to their work, often driven by direct patient care. He expressed concerns about the current process for NIH grants, which may fail to support early-stage risk-taking.
Translational work was another crucial aspect Dr. Gottlieb emphasized, highlighting the mundane yet vital challenges of scaling up manufacturing processes. He pointed to the need for a clear regulatory framework, which has been more readily established in oncology (with its depth of cancer cell biology) compared to other medical areas.
Dr. Gottlieb concluded his talk with optimism, noting the significant strides made in cancer research, particularly in genomics and minimal residual disease (MRD) monitoring. He highlighted the importance of thinking differently in regulatory decision-making, citing instances where unconventional approaches led to favorable outcomes.
####
Following Dr. Gottlieb's presentation, John O'Brien led an insightful interview that touched upon various aspects of healthcare policy, including reimbursement innovation, regulatory flexibility, and the impact of data on healthcare decision-making.
Competitive Bidding for Part B Drugs: Dr. Gottlieb expressed his belief that the entire reimbursement scheme for Part B drugs should be subject to competitive bidding. He argued that Medicare's role as a price taker, combined with its aim to control utilization, raises concerns. He cited the example of the controversial Alzheimer's drug, Aduhelm, where CMS imposed a Coverage with Evidence Development (CED) requirement, effectively denying coverage. He believed that CMS's clinical criteria were flawed and that this approach could have far-reaching consequences.
Challenges of Applying Clinical Criteria: Dr. Gottlieb highlighted a recurring issue where CMS has applied clinical criteria to major coverage decisions and often made incorrect assessments. He gave examples of past cases where CMS's clinical criteria led to suboptimal decisions, such as the use of implantable defibrillators and vascular-delivered valve replacements. He suggested that this approach might be extended to oncology, raising concerns in the field.
Implications of IRA (Innovative Reimbursement Arrangement): Dr. Gottlieb discussed IRA, a mechanism intended to create a synthetic loss of exclusivity for drugs after nine years. He expressed concerns about the impact on small molecular drugs and the potential disincentives for investment. He also noted that the growing elderly population, primarily under Medicare, might face the brunt of these changes. He predicted that reimbursement might be safer for large molecular drugs, and biosimilars could continue their slow price inroads.
Interchangeability and Biosimilars: The conversation touched on the challenges surrounding biosimilar interchangeability. Dr. Gottlieb pointed out the lack of a clear pathway for biosimilars to achieve interchangeability in the market. He suggested that more could be done in terms of interchangeability, emphasizing the importance of legislation and pharmacists' role in switching medications based on an AB rating.
VALID Framework and Regulatory Approaches: The topic of the FDA's recent proposal to regulated LDTs according to the 510(k) and PMA rules. Dr. Gottlieb discussed the VALID legislative framework, supported last year by the FDA's leadership.
The Impact of Data and Regulatory Framework for AI
Dr. Gottlieb and John O'Brien also explored the role of data and regulatory frameworks in the era of artificial intelligence (AI) in healthcare:
Walled Garden Approach to AI: Dr. Gottlieb discussed the FDA's confidence in AI applications that operate on "walled garden" data sets [my term- BQ]. These represent AI tools trained on specific and curated datasets with well-defined outcomes, such as recognizing labeled heart rhythms or differentiating benign from malignant conditions on validated MRI scans. FDA regulators are more comfortable with these applications as they involve clear data and outcomes.
Challenges of Language Models (LLMs): Dr. Gottlieb acknowledged the potential of Language Models (LLMs) for patient engagement and natural language processing but raised concerns about their regulatory complexities. He noted that regulators need guidelines on the datasets used to train LLMs, which currently lack standardization. While LLMs offer unique patient interactions, their regulatory oversight remains a challenge.
From VALID to AI: Interesting Projection. Gottlieb indicated that if the novel VALID laboratory paradigm becomes law, one might use some similar constructs for AI. He explained that the FDA could regulate the firms developing AI tools rather than each individual device, making it important to ensure that the data going in and coming out of AI models are scrutinized, without the need to "rip apart" the technology. He used the concrete example of an Apple Watch and its heart rhythm features, which the FDA approved based on its accuracy "outside the box," without trying to "take apart" and regulate the entire Apple watch and operating system.
##
Regarding AI, FDA released an update on its AI approvals on the next day,10/19/23, here. It was covered in detail at STAT as well.